Tutorial de FreeSurfer n.° 8: Análisis de grupo
Descripción general
Los tutoriales anteriores se han centrado en preparar los datos para un análisis de grupo: primero, los datos se preprocesaron utilizando recon-all, con diferentes medidas estructurales calculadas en cada vértice; y segundo, creamos un archivo FSGD y un archivo de contraste que indica qué grupos queremos comparar entre sí.
Si recuerdas un tutorial anterior, recomendé usar la opción qcache al ejecutar recon-all. Esto generará mapas de espesor, volumen y curvatura con diferentes tamaños de suavizado, como kernels de 0 mm, 10 mm y 25 mm de ancho completo y mitad del máximo. Una de las ventajas del análisis basado en superficies es que permite usar kernels de suavizado mucho más grandes que en los análisis volumétricos, ya que no hay riesgo de suavizado a lo largo de las circunvoluciones. Al ejecutar el análisis de grupo, se puede elegir entre cualquiera de los siguientes tamaños de suavizado:

Ejemplo de salida de qcache. Observe cómo las estimaciones de grosor en los sujetos se suavizan gracias al uso de kernels de suavizado grandes.
Creando un archivo de grupo con mris_preproc
Para realizar un análisis de grupo, necesitaremos combinar todos nuestros mapas estructurales individuales en un único conjunto de datos. Esto es similar a combinar volúmenes consecutivos de una resonancia magnética funcional en un único conjunto de datos, como si los volúmenes estuvieran encadenados y dispuestos uno tras otro. (O, dicho de otro modo, las imágenes estructurales se apilan unas sobre otras, como panqueques; o en capas, como nachos. Use la analogía alimentaria que le resulte más útil para recordar este importante punto).

Los datos también se remuestrean a la plantilla fsaverage, que se encuentra en el espacio MNI. Siempre que realizamos cualquier tipo de análisis de grupo (comparación de grupos, análisis de la región de interés, etc.), los datos de cada sujeto deben tener las mismas dimensiones y resolución de vóxel. Olvidar el remuestreo suele provocar errores durante este paso. (Esto también aplica al análisis fMRI).
Realizaremos todos estos pasos con un solo comando: mris_preproc. El comando requiere los siguientes argumentos:
Un archivo FSGD (indicado por la opción
--fsgd);Una plantilla para remuestrear a (
--target);Una indicación de qué hemisferio se va a remuestrear (
--hemi);Una etiqueta para el archivo de salida (
--out).
En este tutorial, también usaremos la opción --cache-in para especificar las imágenes suavizadas que queremos usar en el análisis. Puede elegir cualquiera de las imágenes suavizadas generadas con la opción --qcache con recon-all.
Para hacer el comando más compacto y adaptable a cualquier estudio que desee analizar, utilizaremos bucles for anidados:
#!/bin/tcsh
estudio setenv $argv[1]
para cada hemisferio (izq. der.)
suavizado foreach (10)
foreach meas (volumen espesor)
mris_preproc --fsgd FSGD/{$estudio}.fsgd \
--cache-in {$meas}.fwhm{$smoothing}.fsaverage \
--objetivo fsaverage \
--hemi {$hemi} \
--out {$hemi}.{$medir}.{$estudio}.{$suavizar}.mgh
fin
fin
fin
Copia este código en un script de shell y guárdalo como runMrisPreproc.sh. También puedes descargarlo aquí.`__. Asegúrese de que el script esté en el directorio que contiene todos los directorios del tema (en este caso, Cannabis/FS) y luego ejecútelo escribiendo tcsh runMrisPreproc.sh.
Ajuste del modelo lineal general con mri_glmfit
Ahora que todos los sujetos están concatenados en un único conjunto de datos, podemos ajustar un modelo lineal general con el comando mri_glmfit de FreeSurfer. En este ejemplo, utilizaremos las siguientes entradas:
El conjunto de datos concatenados que contiene todos los mapas estructurales de los sujetos (
--y);El archivo FSGD (
--fsgd);Una lista de contrastes (cada contraste especificado por una línea diferente que contiene
--C);El hemisferio de la plantilla a analizar (
--surf);Una máscara para restringir nuestro análisis sólo a la corteza (
--cortex);Una etiqueta de salida para el directorio que contiene los resultados (
--glmdir).
Como se indicó anteriormente, utilizaremos bucles for anidados para analizar los hemisferios, los núcleos de suavizado y las medidas estructurales que elijamos. En este ejemplo, analizaremos los hemisferios izquierdo y derecho con un núcleo de suavizado de 10 mm y analizaremos los mapas estructurales de volumen y espesor:
#!/bin/tcsh
conjunto estudio = $argv[1]
para cada hemisferio (izq. der.)
suavidad para cada uno (10)
foreach meas (volumen espesor)
resonancia magnética_glmfit \
--y {$hemi}.{$medida}.{$estudio}.{$suavidad}.mgh \
--fsgd FSGD/{$estudio}.fsgd \
--C Contrastes/CB-HC.mtx \
--C Contrastes/HC-CB.mtx \
--surf fsaverage {$hemi} \
--corteza \
--glmdir {$hemi}.{$medición}.{$estudio}.{$suavidad}.glmdir
fin
fin
fin
- Copia este código en un script de shell y guárdalo como
runGLMs.sh. También puedes descargarlo aquí. __. Ejecute el script desde el directorio del tema escribiendo ``tcsh runGLMs.sh`.
Revisando la salida
If the scripts run without any errors, you should see the following directories in your current directory:
lh.thickness.CannabisStudy.10.glmdir
lh.volume.CannabisStudy.10.glmdir
rh.thickness.CannabisStudy.10.glmdir
rh.volume.CannabisStudy.10.glmdir
The directory name indicates the hemisphere, structural measurement, name of the study, and the smoothing kernel used in the analysis. Each directory contains the same structure; if we navigate into the lh.volume.CannabisStudy.10.glmdir directory and type ls, for example, we will see the following:

The directories CB-HC and HC-CB contain the contrast data for each contrast specified in mri_glmfit, which we will review in the next section. Among the files in the current directory, y.fsgd is a copy of the FSGD file used to run the analysis; mri_glmfit.log contains the code that was run for the current analysis; mask.mgh is the mask used for the analysis; and beta.mgh is a concatenated dataset of the individual beta weights created by the analysis. You can read descriptions of the other outputs by typing mri_glmfit and examining the section under Command-Line Arguments.
Within each of the contrast directories you will see the following:

The file gamma.mgh is a contrast map - the beta dataset in the above directory multiplied by the contrast weights for this particular contrast. In the HC-CB directory, for example, gamma.mgh would show where the Control Group has greater volume than the Cannabis Group in the left hemisphere. z.mgh converts this contrast into a z-map, and sig.mgh converts the contrast into a map of p-values. As above, you can read descriptions of the other outputs in the mri_glmfit help manual.
Note
FreeSurfer uses -log10(p) notation; in other words, a value of 1 in the sig.mgh map represents a p-value of 0.1, a value of 2 represents a p-value of 0.01, and so on.
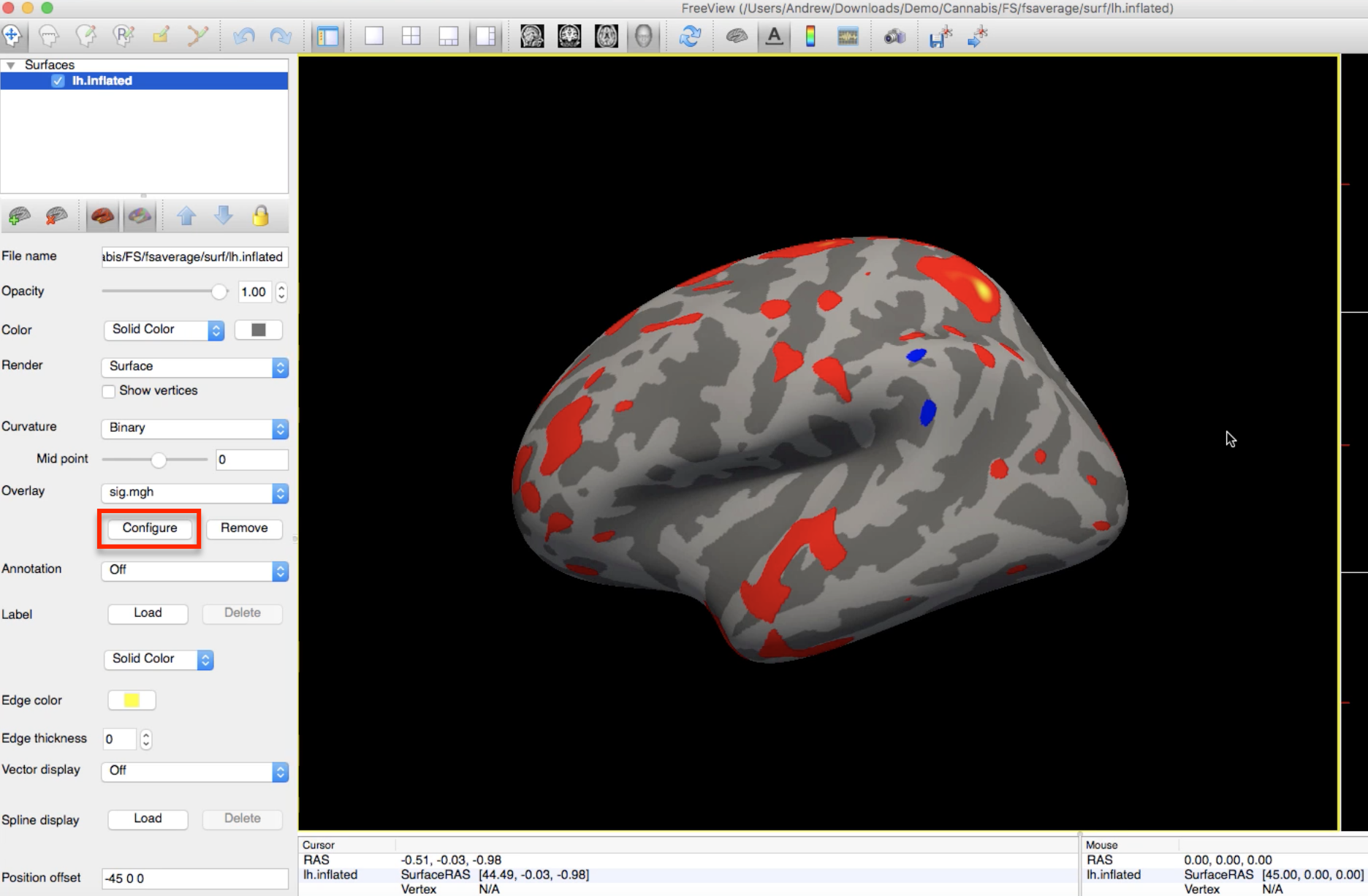
To render the statistical maps on the fsaverage template, navigate to any of the contrast directories (e.g., HC-CB) and type:
freeview -f $SUBJECTS_DIR/fsaverage/surf/lh.inflated:overlay=sig.mgh
Which will load the left hemisphere of the fsaverage template, and map the sig.mgh overlay onto it. The threshold of the p-values can be changed by clicking on the Configure button and moving the sliders.
Now that we have the results, what do we do with them? Your first impulse might be to put them in a manuscript and try to publish it; but to ensure that our results aren’t false positives, we will need to use a form of multiple comparisons correction known as cluster correction, described in the next tutorial.
Video
For a video overview of mris_preproc and mri_glmfit, as well as how to view the results, see `this video
`__.